La stimolazione cerebrale profonda per il trattamento dei disordini del movimento (Parkinson e sindromi distoniche)
Cos’è la malattia di Parkinson? Da quanto tempo è nota? Una breve storia della malattia. Quali sono le sue caratteristiche cliniche e quali i sintomi? Quale evoluzione nel tempo si può prevedere?
La malattia di Parkinson è una delle più frequenti malattie degenerative del sistema nervoso centrale. Già Ippocrate all’epoca degli antichi greci aveva descritto forme di tremore continuo ed inarrestabile. Altri autori alla fine del XVII° secolo descrissero le manifestazioni principali della sindrome parkinsoniana. Tuttavia, la prima descrizione completa della sindrome neurologica è da attribuire a James Parkinson, che in Inghilterra nel 1817 pubblicò una breve monografia, chiamando questa malattia “paralisi agitante”. Egli descrisse “tremori involontari in parti non in movimento, con tendenza a piegare il tronco in avanti e a passare dal camminare al correre, mentre la sensibilità e l’intelligenza sembrano intatte”. In seguito Jean-Martin Charcot, a Parigi nella seconda metà dell’800, approfondì la descrizione, indicando come sintomo cardine la bradicinesia, cioè la lentezza e l’inerzia nell’eseguire i movimenti. E’ a Charcot che si attribuisce il merito di avere sostituito il termine di paralisi agitante con quello di malattia di Parkinson e di avere distinto le due principali forme, la forma tremorigena, cioè dove domina il tremore e la forma rigido acinetica, dove invece domina la rigidità muscolare. Ancora oggi questa distinzione è valida e deve essere considerata anche nella pianificazione della terapia. L’esperienza clinica e l’approfondimento delle conoscenze fisiopatologiche precisarono in seguito le caratteristiche della sindrome: oltre alla bradicinesia il paziente manifesta una marcata rigidità muscolare, tremore, disturbi del controllo della postura e dell’equilibrio. Inoltre è stata riconosciuta una distinzione tra forme a causa identificabile (vascolare, infettiva, tossico-farmacologica), che costituiscono i cosiddetti “parkinsonismi” e forme in cui non è possibile riconoscere un fattore causale, che vengono indicate con il termine di malattia di Parkinson primaria o idiopatica.
All’esordio colpisce soggetti di età media intorno ai 50-60 anni. Esistono forme ad esordio intorno ai 40 anni e forme più tardive dell’anziano. La malattia di Parkinson presenta un’evoluzione clinica per lo più progressiva e difficilmente prevedibile: si può assistere a progressioni lente, che durano molti anni durante i quali le caratteristiche cliniche mutano ed evoluzioni rapide, che portano in poco tempo ad un grave deterioramento motorio ed anche cognitivo fino ad una perdita completa dell’autonomia quotidiana.
Quali armi terapeutiche esistono oggi? Come si sono evolute negli anni?
La terapia di questa malattia ha una storia articolata in varie fasi, chirurgiche e farmacologiche. Fino agli anni ’60 la terapia era prevalentemente chirurgica. Gli interventi chirurgici nei disordini del movimento erano però molto rischiosi, le malattie non erano ancora state ben classificate, le indicazioni non erano standardizzate, la mortalità e i deficit postoperatori troppo elevati. Alla fine degli anni ’40 venne introdotta una tecnica chirurgica, che consentiva di raggiungere un bersaglio all’interno del cranio attraverso una piccola apertura, utilizzando un metodo di puntamento geometrico. Ebbe inizio la “neurochirurgia stereotassica”. In seguito, negli anni ’60, fu introdotto un trattamento farmacologico, che permise di ottenere eccellenti risultati, superiori a quelli chirurgici, senza i rischi di cui questi ultimi erano gravati. Tale terapia medica prevedeva l’impiego di L-Dopa (Levo-Dopa), una sostanza che viene coinvolta nella produzione, all’interno del cervello, di dopamina, un importante neurotrasmettitore. Gli studi di fisiopatologia e di neurochimica hanno permesso di identificare a livello dei nuclei della base la sede delle alterazioni più evidenti, alle quali consegue un marcato deficit di dopamina. Tutt’ora, dopo più di 40 anni, la terapia medica rimane il primo e più importante presidio contro la malattia di Parkinson. Il trattamento con L-Dopa determina un drammatico miglioramento della sintomatologia parkinsoniana, per un periodo di tempo variabile dai 2 ai 10 anni. Purtroppo in seguito compare la cosidetta fase di scompenso della malattia o “sindrome da trattamento a lungo termine con L-DOPA”. Essa è caratterizzata da fluttuazioni della risposta al farmaco con blocchi acinetici e movimenti involontari (discinesie), che risultano molto invalidanti. Possono comparire disturbi del sonno, agitazione fino a sintomi psichiatrici. Oggi il difetto neurochimico può essere contrastato utilizzando precursori della Dopa o farmaci dopamino-agonisti. Successivamente, verso la fine degli anni ’80, si è sviluppata ed è stata applicata all’uomo un’emanazione della neurochirurgia stereotassica, la “neurochirurgia funzionale”, oggi al centro dell’attenzione in neurochirurgia e neurologia, per le concrete possibilità che offre di modulare il funzionamento del cervello e di controllare i sintomi della malattia. Questa chirurgia si propone di identificare un “punto” nel cervello, un centro nervoso e di raggiungerlo mediante strumenti che sono in grado di modificare lo stato di attività di quel centro, ottenendo un miglioramento dello stato clinico del paziente. Alla fine degli anni ‘80, in Europa, per la prima volta al mondo è stata messa a punto una metodica di “stimolazione” di particolari centri del cervello, ottenendo una nuova fase storica della terapia della malattia di Parkinson.
Quali sono gli interventi moderni? Cosa significa DBS?
Per molto tempo, fino agli anni ’80, la neurochirurgia stereotassica si è affidata esclusivamente a metodiche di lesione (coagulazione), cioè di disattivazione irreversibile di centri nervosi al fine di controllare la sintomatologia. Alcuni risultati clinici favorevoli si potevano ottenere, ma erano temporanei e comportavano un rischio di deficit gravi postoperatori molto elevato ed anche una non trascurabile mortalità. Da alcuni anni è stata introdotta una tecnica chirurgica chiamata DBS (Deep Brain Stimulation-Stimolazione Cerebrale Profonda). In molti centri europei ed americani da diversi anni ormai, sono stati chiariti i notevoli e molteplici vantaggi della DBS rispetto alle metodiche chirurgiche di tipo lesivo: si tratta di una metodica reversibile, adattabile e personalizzabile nel singolo paziente, può essere bilaterale e comporta un minore rischio di deficit neurologici permanenti. Gli svantaggi sono pochi e di minore importanza: costo elevato e necessità di sostituire il generatore di impulsi una volta esaurito.
Come si svolge l’intervento chirurgico? Qual è la sua durata?
L’intervento chirurgico di DBS bilaterale prevede due interventi distinti:
-impianto degli elettrodi intracerebrali profondi nel target identificato in anestesia locale
-introduzione del neurostimolatore e dei collegamenti con i due elettrocateteri in anestesia generale
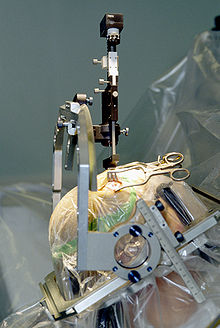
I° intervento. 12 ore prima dell’intervento viene sospesa la terapia medica facendo sì che il paziente entri in sala operatoria con la sua sintomatologia, con fase di blocco e tremori (fase off). In questo modo, essendo i circuiti cerebrali liberi dall’influenza farmacologica, è possibile osservare già in sala operatoria gli effetti della DBS sui segni e sintomi del paziente e gli eventuali effetti collaterali. In anestesia locale praticata a livello frontale ed occipitale viene fissato al capo un anello su cui viene fissato un localizzatore di plexiglas.
Questo sistema forma un “casco”, che include il cervello in un volume in cui si potranno calcolare le coordinate geometriche tridimensionali di ogni suo punto. Il paziente viene trasportato in risonanza magnetica dove viene posto supino con il localizzatore inserito nel supporto del lettino.
Viene sottoposto ad esame NMR e le immagini ottenute vengono inviate ad un computer dotato di un software apposito, che permette di identificare il “bersaglio” (oggi il nucleo subtalamico, STN) e le traiettorie di ingresso in entrambi i lati, per evitare le strutture cerebrali critiche. Si riconduce il paziente in sala operatoria e lo si posiziona sul lettino nella posizione più confortevole possibile, che dovrà essere mantenuta per alcune ore.
Durante tutto l’intervento la collaborazione del paziente è fondamentale, per segnalare effetti collaterali, disturbi e modificazioni cliniche. Per tutta la durata della procedura il paziente non sente dolore e viene avvertito delle fasi più salienti. L’impianto si esegue prima da un lato poi dall’altro. Si identifica un punto di entrata in regione frontale dove si pratica una dose di anestetico locale. L’incisione cutanea è di pochi cm ed il foro è molto piccolo. Si inseriscono delle cannule metalliche, che fungono da guida di sottilissimi microelettrodi e si esegue la fase di registrazione dell’attività dei neuroni.
E’ questa la fase più lunga in quanto i microelettrodi scendono lentamente e ad ogni “fermata” bisogna osservare lo stato di “vitalità” della popolazione neuronale attraversata.
Giunti al bersaglio segue la fase di risalita durante la quale si effettua una stimolazione, per vedere eventuali effetti collaterali. La procedura di registrazione e stimolazione viene effettuata in tutti i microelettrodi fino a che non si identifica la traccia più soddisfacente dal punto di vista neurofisiologico e con gli effetti collaterali più contenuti. Identificata la traccia più idonea, il microelettrodo viene rimosso e sostituito con l’elettrodo definitivo. I contatti stimolanti profondi vengono posizionati a livello delle zone del nucleo in cui si sono ottenute le migliori registrazioni. Nel punto in cui l’elettrodo emerge dall’encefalo si fissa una placchetta di metallo, che terrà ancorato l’elettrodo nella posizione stabilita. La stessa procedura viene ripetuta dal lato opposto. La durata complessiva della procedura è dalle 4 alle 6 ore. Al termine dei due lati gli elettrodi definitivi vengono collegati a due sottili cavi che fuoriescono dalla cute del cranio. Le loro estremità esterne serviranno al neurologo nel periodo di prova per collegarle ad un’apposito stimolatore temporaneo. Questo periodo test dura alcuni giorni e serve a valutare l’effetto clinico, personalizzando il tipo di stimolazione ed evitando effetti collaterali.
II° intervento. Prevede un’anestesia generale. Si esegue una tasca sottocutanea al di sotto della clavicola di dimensioni adeguate per contenere il neurostimolatore definitivo (Kinetra) a destra o a sinistra a seconda delle preferenze del paziente. I cavi che uscivano dal cranio vengono rimossi. Le incisioni frontali vengono riaperte e si collegano i due elettrodi impiantati con due fili adeguatamente isolati, che attraverso il sottocute dal cranio alla clavicola, vengono collegati allo stimolatore. Se necessario si pratica un’incisione intermedia generalmente dietro il padiglione auricolare. Alla fine lo stimolatore è palpabile sotto alla clavicola ma non comporta nessun disturbo. La durata di questo intervento è di 1 ora.
Dove vengono posizionati gli elettrodi? Cos’è il nucleo subtalamico?
Gli elettrodi o elettrocateteri, vengono posizionati all’interno del tessuto cerebrale, con una traiettoria opportunamente predeterminata, indicata dal computer in sala operatoria, per attraversare il cervello evitando strutture funzionali importanti e vasi sanguigni. Il tratto finale dell’elettrodo è costituito da 4 “contatti” metallici, lunghi 1,5 mm con diametro di 1,2 mm dai quali viene erogata la corrente a frequenza, intensità e durata controllate. I contatti sono immersi all’interno di aggregati di cellule cerebrali (neuroni), detti “nuclei”, che nell’encefalo hanno un’anatomia complessa ed una ben precisa organizzazione funzionale. Il nucleo nel quale oggi viene più frequentemente posizionato l’elettrodo è il nucleo subtalamico (NST). Il NST è una struttura densamente popolata (contiene all’incirca 540.000 neuroni), altamente vascolarizzata, situata in una regione profonda del cervello e di dimensioni di pochi mm, quindi per essere “impiantata” necessita di strumenti molto precisi.
Vi sono rischi correlati alla procedura chirurgica?
I rischi specifici legati a questo tipo di intervento chirurgico sono l’emorragia all’interno dell’encefalo e l’infezione dei materiali impiantati. Durante l’intervento è possibile che i sottili vasi cerebrali diano origine ad una raccolta di sangue, che teoricamente può compromettere lo stato neurologico del paziente. Tuttavia, nell’esperienza clinica tale rischio si attesta a valori dello 0,5-2 %, percentuale generica riferibile a tutta la neurochirurgia stereotassica. Le ferite chirurgiche e i materiali che sono stati impiantati possono infettarsi. Generalmente si tratta di infezioni locali che si possono trattare con terapie antibiotiche mirate. Solo raramente è necessario rimuovere lo stimolatore per la presenza di un processo infettivo all’interno della tasca subclaveare.
Quali pazienti parkinsoniani possono trarre beneficio dall’intervento chirurgico? Esistono dei criteri di selezione dei soggetti da sottoporre ad intervento?
L’intervento di DBS prevede una valutazione neurologica preoperatoria, che risponde a rigorosi criteri standardizzati internazionali (CAPSIT-PD: Core Assessment Program for Surgical Interventional Therapies in Parkinson’s Disease). Lo studio preoperatorio è molto articolato e deve comprendere il paziente ed il contesto socio-famigliare in cui egli vive: vita quotidiana, soddisfazione del paziente nei confronti della sua vita attuale, hobby, aspettativa del singolo paziente, ruolo del singolo soggetto nella società, nucleo famigliare e capacità presunta della famiglia di aiutare il soggetto nel postoperatorio. I criteri di inclusione sono i seguenti:
1 - Diagnosi certa di malattia di Parkinson idiopatica. E’ necessario dirimere eventuali dubbi sulla presenza di un parkinsonismo secondario o degerativo atipico. Pazienti affetti da malattia idiopatica e quindi trattabili, che sviluppano nel corso della malattia piccole lesioni vascolari in assenza di elevati fattori di rischio per una malattia cerebrovascolare, non presentano un maggior rischio operatorio e sono selezionabili se rispondono a tutti gli altri criteri. Ogni elemento, clinico o strumentale, anche isolato, che ponga in dubbio la diagnosi di malattia di Parkinson idiopatica, impone un’osservazione e controlli nel tempo prima di fare l’intervento. La malattia deve avere una durata di almeno 5 anni. Bisogna escludere un’atrofia multisistemica, che tuttavia generalmente esordisce bilateralmente mentre il Parkinson esordisce in genere prima da un lato, poi passa all’altro.
2 - Età. Con le riserve dettate dalla valutazione dell’età biologica piuttosto che quella anagrafica, la fascia di età più idonea è 35-70 anni. Tuttavia, già al di sopra dei 65 anni bisogna attentamente valutare le condizioni del paziente, le aspettative ed il rischio operatorio.
3 - Malattia di Parkinson complicata. La gravità della malattia deve essere valutata determinando la presenza di complicanze motorie, principalmente causate dalla terapia farmacologica (da mantenere invariata per almeno 1 mese): fluttuazioni, discinesie e movimenti involontari, non correggibili in modo soddisfacente con le altre strategie farmacologiche disponibili. La stimolazione subtalamica è molto efficace nel ridurre i tremori gravi insostenibili. Il STN è dotato anche di un effetto potente nei confronti delle distonie dolorose in fase off, dove si pone una specifica indicazione.
4 - Grado di disabilità. La disabilità motoria, cioè comparsa dei sintomi dopo almeno 12 ore di sospensione della terapia, secondo scale apposite, deve essere severa, tale da richiedere un’assistenza nella maggior parte delle attività quotidiane.
5 - Entità della risposta alla L-Dopa. Il paziente deve riferire la conservazione di una buona risposta alla L-Dopa. Prima dell’intervento si esegue un test per valutare tale sensibilità al farmaco.
6 - Aspetto psichiatrico. In anamnesi devono essere assenti patologie come depressione e psicosi maggiori. Viene tollerata un’ansia lieve e/o una depressione modesta e reattiva. Le eventuali psicosi da farmaci pregresse devono essere risolte con dosi moderate di neurolettici atipici. La risoluzione deve essere accertata chiedendo esplicitamente al paziente se presenta ancora le allucinazioni. Se il farmaco neurolettico deve essere usato cronicamente per controllare il disturbo il paziente viene escluso. In assenza di un trattamento con neurolettico, possono essere accettate rare allucinazioni benigne, che possono essere correlate ai farmaci. Questo perché, come tali, potrebbero essere risolte con la DBS, che consente una netta riduzione della terapia farmacologica.
7 - Caratteristiche psicologiche, comportamentali e neuropsicologiche. Il paziente deve offrire una adeguata collaborazione. Deve essere in grado di fornire una collaborazione terapeutica accettabile rispettando le prescrizioni. Deve eseguire controlli frequenti, di aggiustamenti della stimolazione profonda e attendere un periodo di tempo di diversi mesi, durante il quale si verificherà l’adattamento dei circuiti alla stimolazione. Anche i famigliari devono essere collaboranti e aiutare il paziente nella gestione dello stimolatore. Vengono esclusi pazienti con demenza e con gravi deficit dei lobi frontali.
8 - Valutazione neuroradiologica. Si esegue sempre una NMR cerebrale prima dell’intervento chirurgico. E’ l’esame diagnostico che fornisce il maggior numero di dettagli anatomici per decidere se operare il paziente. L’indagine deve risultare “normale” o presentare al massimo segni “modesti” di encefalopatia vascolare.
9 - Valutazione neurofisiologica ed internistica generale. Di fatto non esistono controindicazioni specifiche all’intervento. Il soggetto viene inquadrato da un punto di vista internistico per valutare l’eventuale presenza di patologie sistemiche importanti (tumori maligni, diabete grave, ecc), che costituirebbero un fattore di rischio operatorio o un fattore prognosticamente negativo in ogni tipo di intervento chirurgico. In particolare vengono indagati l’assetto cardio-circolatorio e metabolico. Queste valutazioni comprendono anche l’insieme di indagini neurofisiologiche mirate alla valutazione dell’integrità funzionale del cervello (PESS, PEV, PEM).
La selezione dei pazienti è un compito che spetta principalmente al neurologo. Il neurochirurgo, individuato un possibile candidato, viene coinvolto nella fase di consenso informato, per esporre i benefici e i rischi inerenti l’intervento chirurgico.
Cos’è la distonia? Che età può interessare?
Le distonie o più correttamente “sindromi distoniche” sono un gruppo molto eterogeneo di quadri clinici, eziopatogenetici e fisiopatologici. Sono malattie caratterizzate da movimenti involontari, che derivano da una errata coordinazione tra i vari gruppi muscolari del corpo. Ne risultano dei movimenti (distonie mobili) e delle posizioni o posture anomale (distonie fisse), completamente svincolate dalla finalità specifica del movimento stesso. Il paziente può avere continui movimenti incontrollati, che rendono molto difficoltosa o del tutto inaccettabile la vita di relazione. Possono interessare sia l’età infantile, che l’età adulta. Distinguiamo forme secondarie, conseguenti a lesioni cerebrali di natura vascolare, traumatica, tossico-metabolica, ecc. , comunque nota e forme primarie, idiopatiche, correlate a numerose mutazioni genetiche, che sono solo in parte note. Distinguiamo inoltre forme generalizzate, che interessano tutto il corpo e forme focali, che coinvolgono solo certe parti, come gli arti o il tronco. Negli anni il soggetto distonico continuamente interessato da movimenti complessi e non finalizzati sviluppa gravi complicanze osteo-muscolari, che fissano tronco e arti in posizioni e posture viziate, che non sono più risolvibili anche riducendo i movimenti involontari.
Qual è l’origine e come si sviluppa la malattia?
Sebbene appare chiaro che la comparsa della sintomatologia distonica consegua a danni più o meno precoci nell’attività dei neuroni dei circuiti cerebrali più profondi (detti circuiti pallido-talamo-corticali), l’esatta relazione tra tali anomalie e le manifestazioni della malattia rimane non chiara. Gli studi non hanno ancora definito un meccanismo fisiopatologico univoco. Le alterazioni dei circuiti tra corteccia e nuclei profondi sono comunque molto variabili e dipendono molto dal tipo di malattia alla base della sindrome distonica. Talvolta i centri nervosi coinvolti sono iperattivi, cioè funzionano di più rispetto al soggetto normale, talvolta mostrano invece un’ipoattivazione, cioè sono meno attivi, con alcune similitudini rispetto alla malattia di Parkinson.
Quali terapie sono possibili?
L’avvento della neurochirurgia stereotassica funzionale ha segnato un momento fondamentale anche per il trattamento delle distonie. Fino al 1990 le procedure stereotassiche per trattare le forme distoniche avevano come target prevalente un centro, il “globo pallido” (Gpi), dove si praticavano lesioni. L’intervento era detto “pallidotomia” ed era irreversibile e rischioso. In seguito è stata introdotta la DBS anche per il trattamento delle sindromi distoniche. Nell’arco di pochi anni la DBS si è largamente diffusa ed il centro nervoso interessato è rimasto lo stesso. La terapia farmacologica non ha dato grossi benefici, sono state impiegate sostanza generiche, attive sul sistema nervoso centrale, già usate in molte altre malattie nervose. In altre parole, una sostanza come era stata la “L-Dopa” per la malattia di Parkinson, utile nella stessa misura per il trattamento delle distonie, non è stata mai individuata. Molte forme sono resistenti alla terapia medica e si instaurano terapie croniche soltanto sintomatiche, peraltro non scevre di effetti collaterali.
Quali risultati può ottenere la DBS? Quali sono i rischi?
Gli eccellenti risultati ottenuti nel trattamento della distonia generalizzata idiopatica resistente alla terapia medica ed anche di altre forme più focali, come quelle cervicali, ha consolidato la procedura di DBS del globo pallido come trattamento di scelta. Nelle casistiche maggiori vengono descritti miglioramenti dei punteggi nelle scale di valutazione della distonia di più dell’80 %. Tale valore, riferito a tutti i soggetti operati, si innalza al 90,3 % in certi sottogruppi di distonie, come la PGD (Primary Generalized Dystonia)-DYT1 positiva, cioè positiva per un ben preciso gene nel cromosoma 9, che è stato isolato e codificato. Ciò supporta la validità della tecnica chirurgica e del target in tutte le forme idiopatiche e soprattutto nelle forme a trasmissione ereditaria.
Nelle distonie mobili, che interessano soprattutto gli arti in movimento, il miglioramento clinico postoperatorio può essere precoce, fin dai primi giorni. Le distonie fisse invece, a carico del tronco e a riposo, presentano un miglioramento più tardivo, anche dopo mesi dall’intervento. Comunque, per valutare i risultati clinici dopo l’intervento è necessario attendere diversi mesi. Infatti anche se molti autori hanno descritto un decremento evidente dei movimenti involontari anormali subito dopo l’accensione dello stimolatore, il beneficio sulle posture e sull’aspetto funzionale globale si può valutare solo a distanza.
Dopo l’impianto è anche possibile intervenire sulle deformità secondarie dell’apparato muscolo-scheletrico, talora molto deturpanti e che, come già detto, spesso sono irreversibili anche a fronte del miglioramento neurologico ottenuto. Questo aspetto depone a favore di una necessaria precocità dell’intervento chirurgico. La procedura deve essere considerata fin dalle fasi iniziali della malattia, specie nei bambini, per i buoni risultati clinici ottenibili nella maggior parte dei pazienti, per la mancanza di un’alternativa medica appropriata, per la caratteristica di progressione nel tempo della malattia e per le gravi complicanze ortopediche che determina. Il paziente prima di essere operato viene studiato dal neurologo in modo approfondito. Esistono dei protocolli valutativi pre- e post- chirurgici molto dettagliati (valutazione clinica, neurofisiologica e molecolare), che analizzano tutti gli aspetti delle vita quotidiana del paziente. Il trattamento della distonia ad esordio precoce in età evolutiva è infatti qualcosa che coinvolge attivamente tutta la famiglia. Per quanto riguarda i rischi chirurgici specifici si rimanda alle considerazioni fatte per la chirurgia della malattia di Parkinson, inerenti le infezioni dei materiali impiantati e le emorragie cerebrali.
Qual è lo studio pre-chirurgico di selezione a cui tutti i soggetti devono sottoporsi?
1 - Esame neurologico finalizzato a valutare il controllo del capo, il controllo posturale, la persistenza di riflessi, la presenza di schemi patologici, il grado di ipertonia, l’eventuale presenza di clonie, la selettività dei movimenti, i passaggi posturali, le reazioni di equilibrio, la presa ecc.
2 - Studio neurofisiologico mediante EEG-poligrafia in corso di veglia e di sonno spontaneo con attivazioni di routine, BAEP, PESS e in casi selezionati ENG, EMG.
3 - Studio di neuroimaging mediante NMR dell’encefalo. Nella maggioranza dei casi l’esame NMR è negativo.
4 - Studio biochimico del sangue per valutare alcune sostanze che sono diagnostiche per certe malattie neurologiche. Oltre a una normale routine ematochimica vengono dosate CPK, LDH, lattato, ceruloplasmina, cupremia, aldolasi, uricemia. In casi selezionati si eseguono anche il dosaggio degli acidi organici urinari e delle acil-carnitine urinarie, acantociti, alfafetoproteina, IgA, IgE, enzimi lisosomiali (arisulfatasi A, esosaminidasi A e B, beta galattosidasi), dosaggio della neopterina in linfoblasti.
5 - Studio molecolare per l’identificazione dei geni e delle mutazioni. La più nota è la delezione GAG nel gene DYT-1 responsabile della distonia idiopatica generalizzata di torsione (Oppenheim dystonia), già precedentemente menzionata. Inoltre, in casi selezionati (pazienti per i quali, sulla base dei dati clinici, strumentali e bioumorali, venga sospettata una possibile patologia mitocondriale) vengono ricercate le mutazioni del mtDNA più frequentemente coinvolte nei quadri encefalopatici
6 - Scala per la valutazione della distonia (Burke-Fahn-Mardsen, BFM). La scala serve a valutare i movimenti distonici e le posture distoniche. Comprende l’osservazione diretta del soggetto e appositi video (osservazione a riposo, movimenti involontari, lettura e fonazione, prove di scrittura, prove di abilità manuale, ecc). La scala conferisce un punteggio massimo di 120. Inoltre si eseguono scale di disabilità per linguaggio, scrittura, alimentazione, nutrizione, igiene, deambulazione, vestirsi.
7 - Intervista sulla disabilità riguardante l’autonomia nella cura di sé, somministrata ai genitori del paziente.
8 - Profilo neuropsicologico con valutazione delle funzioni cognitive generali, delle funzioni visuospaziali, esecutive, mnesiche e percettive.
9 - Valutazione logopedica adeguata e bilancio della capacità di esprimersi verbalmente.
Tutti i pazienti vengono sottoposti ad un tentativo farmacologico con L-Dopa, per poter identificare eventuali forme di distonia responsiva a questa sostanza, che sono comunque relativamente rare. Viene inoltre intrapreso un trattamento farmacologico mediante l’uso isolato oppure l’associazione di L-Dopa, Anticolinergici, Tetrabenazina o Baclofen.
Esistono criteri di inclusione per la DBS?
In base al protocollo di studio preoperatorio descritto, possiamo schematizzarli come segue:
1 - età superiore a 7 anni
2 - NMR cerebrale negativa
3 - profilo cognitivo-psichiatrico normale e adeguato screening preoperatorio
4 - assenza di risposta clinicamente rilevante al trattamento farmacologico
5 - disabilità severa, fortemente condizionata dalla componente distonica (BFM > 50-60)
Come si svolge l’intervento neurochirurgico?
La procedura di DBS, quando viene eseguita in paziente adulti, prevede il paziente sveglio e la microregistrazione intraoperatoria. La metodica è analoga al trattamento della malattia di Parkinson. Nei bambini invece si impiega un’anestesia generale con o senza microregistrazione (
In età pediatrica si impianta tutto lo stesso giorno, sia gli elettrocateteri cerebrali, che lo stimolatore definitivo, mentre negli adulti si praticano due interventi distinti, come nel Parkinson. Dopo una settimana dall’impianto si procede all’accensione dello stimolatore ad alta frequenza, con intensità maggiori rispetto al Parkinson, che vegono progressivamente incrementate a seconda dei risultati clinici.
Come viene seguito il paziente operato? In questa indicazione chirurgica relativamente nuova, alla luce dell’attuale esperienza quali considerazioni si possono fare?
Dopo la dimissione dalla Neurochirurgia i pazienti vengono seguiti clinicamente dal neurologo con scadenze settimanali per il primo mese ed, in seguito con controlli sequenziali a 1, 3, 6, 12 mesi dall’intervento. Vengono applicati gli stessi test valutativi somministrati in fase pre intervento. Contemporaneamente all’accensione dello stimolatore si procede al trattamento riabilitativo (logopedia, terapia occupazionale, fisiochinesiterapia) volto a modellare i nuovi schemi motori esistenti, sulla base di maggiori capacità di controllo del movimento e della postura.
La selezione dei pazienti distonici è fondamentale per ottenere il maggior beneficio clinico e soggettivo da un intervento di DBS, minimizzando gli effetti avversi, sia nelle forme primarie che secondarie. Vi è un crescente consenso nel ritenere che l’intervento dovrebbe pertanto essere proposto come trattamento di prima linea nei casi di distonia generalizzata primaria e secondaria non responsiva alla terapia farmacologica.
Cos’è la malattia di Parkinson? Da quanto tempo è nota? Una breve storia della malattia. Quali sono le sue caratteristiche cliniche e quali i sintomi? Quale evoluzione nel tempo si può prevedere?
La malattia di Parkinson è una delle più frequenti malattie degenerative del sistema nervoso centrale. Già Ippocrate all’epoca degli antichi greci aveva descritto forme di tremore continuo ed inarrestabile. Altri autori alla fine del XVII° secolo descrissero le manifestazioni principali della sindrome parkinsoniana. Tuttavia, la prima descrizione completa della sindrome neurologica è da attribuire a James Parkinson, che in Inghilterra nel 1817 pubblicò una breve monografia, chiamando questa malattia “paralisi agitante”. Egli descrisse “tremori involontari in parti non in movimento, con tendenza a piegare il tronco in avanti e a passare dal camminare al correre, mentre la sensibilità e l’intelligenza sembrano intatte”. In seguito Jean-Martin Charcot, a Parigi nella seconda metà dell’800, approfondì la descrizione, indicando come sintomo cardine la bradicinesia, cioè la lentezza e l’inerzia nell’eseguire i movimenti. E’ a Charcot che si attribuisce il merito di avere sostituito il termine di paralisi agitante con quello di malattia di Parkinson e di avere distinto le due principali forme, la forma tremorigena, cioè dove domina il tremore e la forma rigido acinetica, dove invece domina la rigidità muscolare. Ancora oggi questa distinzione è valida e deve essere considerata anche nella pianificazione della terapia. L’esperienza clinica e l’approfondimento delle conoscenze fisiopatologiche precisarono in seguito le caratteristiche della sindrome: oltre alla bradicinesia il paziente manifesta una marcata rigidità muscolare, tremore, disturbi del controllo della postura e dell’equilibrio. Inoltre è stata riconosciuta una distinzione tra forme a causa identificabile (vascolare, infettiva, tossico-farmacologica), che costituiscono i cosiddetti “parkinsonismi” e forme in cui non è possibile riconoscere un fattore causale, che vengono indicate con il termine di malattia di Parkinson primaria o idiopatica.
All’esordio colpisce soggetti di età media intorno ai 50-60 anni. Esistono forme ad esordio intorno ai 40 anni e forme più tardive dell’anziano. La malattia di Parkinson presenta un’evoluzione clinica per lo più progressiva e difficilmente prevedibile: si può assistere a progressioni lente, che durano molti anni durante i quali le caratteristiche cliniche mutano ed evoluzioni rapide, che portano in poco tempo ad un grave deterioramento motorio ed anche cognitivo fino ad una perdita completa dell’autonomia quotidiana.
Quali armi terapeutiche esistono oggi? Come si sono evolute negli anni?
La terapia di questa malattia ha una storia articolata in varie fasi, chirurgiche e farmacologiche. Fino agli anni ’60 la terapia era prevalentemente chirurgica. Gli interventi chirurgici nei disordini del movimento erano però molto rischiosi, le malattie non erano ancora state ben classificate, le indicazioni non erano standardizzate, la mortalità e i deficit postoperatori troppo elevati. Alla fine degli anni ’40 venne introdotta una tecnica chirurgica, che consentiva di raggiungere un bersaglio all’interno del cranio attraverso una piccola apertura, utilizzando un metodo di puntamento geometrico. Ebbe inizio la “neurochirurgia stereotassica”. In seguito, negli anni ’60, fu introdotto un trattamento farmacologico, che permise di ottenere eccellenti risultati, superiori a quelli chirurgici, senza i rischi di cui questi ultimi erano gravati. Tale terapia medica prevedeva l’impiego di L-Dopa (Levo-Dopa), una sostanza che viene coinvolta nella produzione, all’interno del cervello, di dopamina, un importante neurotrasmettitore. Gli studi di fisiopatologia e di neurochimica hanno permesso di identificare a livello dei nuclei della base la sede delle alterazioni più evidenti, alle quali consegue un marcato deficit di dopamina. Tutt’ora, dopo più di 40 anni, la terapia medica rimane il primo e più importante presidio contro la malattia di Parkinson. Il trattamento con L-Dopa determina un drammatico miglioramento della sintomatologia parkinsoniana, per un periodo di tempo variabile dai 2 ai 10 anni. Purtroppo in seguito compare la cosidetta fase di scompenso della malattia o “sindrome da trattamento a lungo termine con L-DOPA”. Essa è caratterizzata da fluttuazioni della risposta al farmaco con blocchi acinetici e movimenti involontari (discinesie), che risultano molto invalidanti. Possono comparire disturbi del sonno, agitazione fino a sintomi psichiatrici. Oggi il difetto neurochimico può essere contrastato utilizzando precursori della Dopa o farmaci dopamino-agonisti. Successivamente, verso la fine degli anni ’80, si è sviluppata ed è stata applicata all’uomo un’emanazione della neurochirurgia stereotassica, la “neurochirurgia funzionale”, oggi al centro dell’attenzione in neurochirurgia e neurologia, per le concrete possibilità che offre di modulare il funzionamento del cervello e di controllare i sintomi della malattia. Questa chirurgia si propone di identificare un “punto” nel cervello, un centro nervoso e di raggiungerlo mediante strumenti che sono in grado di modificare lo stato di attività di quel centro, ottenendo un miglioramento dello stato clinico del paziente. Alla fine degli anni ‘80, in Europa, per la prima volta al mondo è stata messa a punto una metodica di “stimolazione” di particolari centri del cervello, ottenendo una nuova fase storica della terapia della malattia di Parkinson.
Quali sono gli interventi moderni? Cosa significa DBS?
Per molto tempo, fino agli anni ’80, la neurochirurgia stereotassica si è affidata esclusivamente a metodiche di lesione (coagulazione), cioè di disattivazione irreversibile di centri nervosi al fine di controllare la sintomatologia. Alcuni risultati clinici favorevoli si potevano ottenere, ma erano temporanei e comportavano un rischio di deficit gravi postoperatori molto elevato ed anche una non trascurabile mortalità. Da alcuni anni è stata introdotta una tecnica chirurgica chiamata DBS (Deep Brain Stimulation-Stimolazione Cerebrale Profonda). In molti centri europei ed americani da diversi anni ormai, sono stati chiariti i notevoli e molteplici vantaggi della DBS rispetto alle metodiche chirurgiche di tipo lesivo: si tratta di una metodica reversibile, adattabile e personalizzabile nel singolo paziente, può essere bilaterale e comporta un minore rischio di deficit neurologici permanenti. Gli svantaggi sono pochi e di minore importanza: costo elevato e necessità di sostituire il generatore di impulsi una volta esaurito.
Come si svolge l’intervento chirurgico? Qual è la sua durata?
L’intervento chirurgico di DBS bilaterale prevede due interventi distinti:
-impianto degli elettrodi intracerebrali profondi nel target identificato in anestesia locale
-introduzione del neurostimolatore e dei collegamenti con i due elettrocateteri in anestesia generale
I° intervento. 12 ore prima dell’intervento viene sospesa la terapia medica facendo sì che il paziente entri in sala operatoria con la sua sintomatologia, con fase di blocco e tremori (fase off). In questo modo, essendo i circuiti cerebrali liberi dall’influenza farmacologica, è possibile osservare già in sala operatoria gli effetti della DBS sui segni e sintomi del paziente e gli eventuali effetti collaterali. In anestesia locale praticata a livello frontale ed occipitale viene fissato al capo un anello su cui viene fissato un localizzatore di plexiglas.
Questo sistema forma un “casco”, che include il cervello in un volume in cui si potranno calcolare le coordinate geometriche tridimensionali di ogni suo punto. Il paziente viene trasportato in risonanza magnetica dove viene posto supino con il localizzatore inserito nel supporto del lettino.
Viene sottoposto ad esame NMR e le immagini ottenute vengono inviate ad un computer dotato di un software apposito, che permette di identificare il “bersaglio” (oggi il nucleo subtalamico, STN) e le traiettorie di ingresso in entrambi i lati, per evitare le strutture cerebrali critiche. Si riconduce il paziente in sala operatoria e lo si posiziona sul lettino nella posizione più confortevole possibile, che dovrà essere mantenuta per alcune ore.
Durante tutto l’intervento la collaborazione del paziente è fondamentale, per segnalare effetti collaterali, disturbi e modificazioni cliniche. Per tutta la durata della procedura il paziente non sente dolore e viene avvertito delle fasi più salienti. L’impianto si esegue prima da un lato poi dall’altro. Si identifica un punto di entrata in regione frontale dove si pratica una dose di anestetico locale. L’incisione cutanea è di pochi cm ed il foro è molto piccolo. Si inseriscono delle cannule metalliche, che fungono da guida di sottilissimi microelettrodi e si esegue la fase di registrazione dell’attività dei neuroni.
E’ questa la fase più lunga in quanto i microelettrodi scendono lentamente e ad ogni “fermata” bisogna osservare lo stato di “vitalità” della popolazione neuronale attraversata.
Giunti al bersaglio segue la fase di risalita durante la quale si effettua una stimolazione, per vedere eventuali effetti collaterali. La procedura di registrazione e stimolazione viene effettuata in tutti i microelettrodi fino a che non si identifica la traccia più soddisfacente dal punto di vista neurofisiologico e con gli effetti collaterali più contenuti. Identificata la traccia più idonea, il microelettrodo viene rimosso e sostituito con l’elettrodo definitivo. I contatti stimolanti profondi vengono posizionati a livello delle zone del nucleo in cui si sono ottenute le migliori registrazioni. Nel punto in cui l’elettrodo emerge dall’encefalo si fissa una placchetta di metallo, che terrà ancorato l’elettrodo nella posizione stabilita. La stessa procedura viene ripetuta dal lato opposto. La durata complessiva della procedura è dalle 4 alle 6 ore. Al termine dei due lati gli elettrodi definitivi vengono collegati a due sottili cavi che fuoriescono dalla cute del cranio. Le loro estremità esterne serviranno al neurologo nel periodo di prova per collegarle ad un’apposito stimolatore temporaneo. Questo periodo test dura alcuni giorni e serve a valutare l’effetto clinico, personalizzando il tipo di stimolazione ed evitando effetti collaterali.
II° intervento. Prevede un’anestesia generale. Si esegue una tasca sottocutanea al di sotto della clavicola di dimensioni adeguate per contenere il neurostimolatore definitivo (Kinetra) a destra o a sinistra a seconda delle preferenze del paziente. I cavi che uscivano dal cranio vengono rimossi. Le incisioni frontali vengono riaperte e si collegano i due elettrodi impiantati con due fili adeguatamente isolati, che attraverso il sottocute dal cranio alla clavicola, vengono collegati allo stimolatore. Se necessario si pratica un’incisione intermedia generalmente dietro il padiglione auricolare. Alla fine lo stimolatore è palpabile sotto alla clavicola ma non comporta nessun disturbo. La durata di questo intervento è di 1 ora.
Dove vengono posizionati gli elettrodi? Cos’è il nucleo subtalamico?
Gli elettrodi o elettrocateteri, vengono posizionati all’interno del tessuto cerebrale, con una traiettoria opportunamente predeterminata, indicata dal computer in sala operatoria, per attraversare il cervello evitando strutture funzionali importanti e vasi sanguigni. Il tratto finale dell’elettrodo è costituito da 4 “contatti” metallici, lunghi 1,5 mm con diametro di 1,2 mm dai quali viene erogata la corrente a frequenza, intensità e durata controllate. I contatti sono immersi all’interno di aggregati di cellule cerebrali (neuroni), detti “nuclei”, che nell’encefalo hanno un’anatomia complessa ed una ben precisa organizzazione funzionale. Il nucleo nel quale oggi viene più frequentemente posizionato l’elettrodo è il nucleo subtalamico (NST). Il NST è una struttura densamente popolata (contiene all’incirca 540.000 neuroni), altamente vascolarizzata, situata in una regione profonda del cervello e di dimensioni di pochi mm, quindi per essere “impiantata” necessita di strumenti molto precisi.
Vi sono rischi correlati alla procedura chirurgica?
I rischi specifici legati a questo tipo di intervento chirurgico sono l’emorragia all’interno dell’encefalo e l’infezione dei materiali impiantati. Durante l’intervento è possibile che i sottili vasi cerebrali diano origine ad una raccolta di sangue, che teoricamente può compromettere lo stato neurologico del paziente. Tuttavia, nell’esperienza clinica tale rischio si attesta a valori dello 0,5-2 %, percentuale generica riferibile a tutta la neurochirurgia stereotassica. Le ferite chirurgiche e i materiali che sono stati impiantati possono infettarsi. Generalmente si tratta di infezioni locali che si possono trattare con terapie antibiotiche mirate. Solo raramente è necessario rimuovere lo stimolatore per la presenza di un processo infettivo all’interno della tasca subclaveare.
Quali pazienti parkinsoniani possono trarre beneficio dall’intervento chirurgico? Esistono dei criteri di selezione dei soggetti da sottoporre ad intervento?
L’intervento di DBS prevede una valutazione neurologica preoperatoria, che risponde a rigorosi criteri standardizzati internazionali (CAPSIT-PD: Core Assessment Program for Surgical Interventional Therapies in Parkinson’s Disease). Lo studio preoperatorio è molto articolato e deve comprendere il paziente ed il contesto socio-famigliare in cui egli vive: vita quotidiana, soddisfazione del paziente nei confronti della sua vita attuale, hobby, aspettativa del singolo paziente, ruolo del singolo soggetto nella società, nucleo famigliare e capacità presunta della famiglia di aiutare il soggetto nel postoperatorio. I criteri di inclusione sono i seguenti:
1 - Diagnosi certa di malattia di Parkinson idiopatica. E’ necessario dirimere eventuali dubbi sulla presenza di un parkinsonismo secondario o degerativo atipico. Pazienti affetti da malattia idiopatica e quindi trattabili, che sviluppano nel corso della malattia piccole lesioni vascolari in assenza di elevati fattori di rischio per una malattia cerebrovascolare, non presentano un maggior rischio operatorio e sono selezionabili se rispondono a tutti gli altri criteri. Ogni elemento, clinico o strumentale, anche isolato, che ponga in dubbio la diagnosi di malattia di Parkinson idiopatica, impone un’osservazione e controlli nel tempo prima di fare l’intervento. La malattia deve avere una durata di almeno 5 anni. Bisogna escludere un’atrofia multisistemica, che tuttavia generalmente esordisce bilateralmente mentre il Parkinson esordisce in genere prima da un lato, poi passa all’altro.
2 - Età. Con le riserve dettate dalla valutazione dell’età biologica piuttosto che quella anagrafica, la fascia di età più idonea è 35-70 anni. Tuttavia, già al di sopra dei 65 anni bisogna attentamente valutare le condizioni del paziente, le aspettative ed il rischio operatorio.
3 - Malattia di Parkinson complicata. La gravità della malattia deve essere valutata determinando la presenza di complicanze motorie, principalmente causate dalla terapia farmacologica (da mantenere invariata per almeno 1 mese): fluttuazioni, discinesie e movimenti involontari, non correggibili in modo soddisfacente con le altre strategie farmacologiche disponibili. La stimolazione subtalamica è molto efficace nel ridurre i tremori gravi insostenibili. Il STN è dotato anche di un effetto potente nei confronti delle distonie dolorose in fase off, dove si pone una specifica indicazione.
4 - Grado di disabilità. La disabilità motoria, cioè comparsa dei sintomi dopo almeno 12 ore di sospensione della terapia, secondo scale apposite, deve essere severa, tale da richiedere un’assistenza nella maggior parte delle attività quotidiane.
5 - Entità della risposta alla L-Dopa. Il paziente deve riferire la conservazione di una buona risposta alla L-Dopa. Prima dell’intervento si esegue un test per valutare tale sensibilità al farmaco.
6 - Aspetto psichiatrico. In anamnesi devono essere assenti patologie come depressione e psicosi maggiori. Viene tollerata un’ansia lieve e/o una depressione modesta e reattiva. Le eventuali psicosi da farmaci pregresse devono essere risolte con dosi moderate di neurolettici atipici. La risoluzione deve essere accertata chiedendo esplicitamente al paziente se presenta ancora le allucinazioni. Se il farmaco neurolettico deve essere usato cronicamente per controllare il disturbo il paziente viene escluso. In assenza di un trattamento con neurolettico, possono essere accettate rare allucinazioni benigne, che possono essere correlate ai farmaci. Questo perché, come tali, potrebbero essere risolte con la DBS, che consente una netta riduzione della terapia farmacologica.
7 - Caratteristiche psicologiche, comportamentali e neuropsicologiche. Il paziente deve offrire una adeguata collaborazione. Deve essere in grado di fornire una collaborazione terapeutica accettabile rispettando le prescrizioni. Deve eseguire controlli frequenti, di aggiustamenti della stimolazione profonda e attendere un periodo di tempo di diversi mesi, durante il quale si verificherà l’adattamento dei circuiti alla stimolazione. Anche i famigliari devono essere collaboranti e aiutare il paziente nella gestione dello stimolatore. Vengono esclusi pazienti con demenza e con gravi deficit dei lobi frontali.
8 - Valutazione neuroradiologica. Si esegue sempre una NMR cerebrale prima dell’intervento chirurgico. E’ l’esame diagnostico che fornisce il maggior numero di dettagli anatomici per decidere se operare il paziente. L’indagine deve risultare “normale” o presentare al massimo segni “modesti” di encefalopatia vascolare.
9 - Valutazione neurofisiologica ed internistica generale. Di fatto non esistono controindicazioni specifiche all’intervento. Il soggetto viene inquadrato da un punto di vista internistico per valutare l’eventuale presenza di patologie sistemiche importanti (tumori maligni, diabete grave, ecc), che costituirebbero un fattore di rischio operatorio o un fattore prognosticamente negativo in ogni tipo di intervento chirurgico. In particolare vengono indagati l’assetto cardio-circolatorio e metabolico. Queste valutazioni comprendono anche l’insieme di indagini neurofisiologiche mirate alla valutazione dell’integrità funzionale del cervello (PESS, PEV, PEM).
La selezione dei pazienti è un compito che spetta principalmente al neurologo. Il neurochirurgo, individuato un possibile candidato, viene coinvolto nella fase di consenso informato, per esporre i benefici e i rischi inerenti l’intervento chirurgico.
Cos’è la distonia? Che età può interessare?
Le distonie o più correttamente “sindromi distoniche” sono un gruppo molto eterogeneo di quadri clinici, eziopatogenetici e fisiopatologici. Sono malattie caratterizzate da movimenti involontari, che derivano da una errata coordinazione tra i vari gruppi muscolari del corpo. Ne risultano dei movimenti (distonie mobili) e delle posizioni o posture anomale (distonie fisse), completamente svincolate dalla finalità specifica del movimento stesso. Il paziente può avere continui movimenti incontrollati, che rendono molto difficoltosa o del tutto inaccettabile la vita di relazione. Possono interessare sia l’età infantile, che l’età adulta. Distinguiamo forme secondarie, conseguenti a lesioni cerebrali di natura vascolare, traumatica, tossico-metabolica, ecc. , comunque nota e forme primarie, idiopatiche, correlate a numerose mutazioni genetiche, che sono solo in parte note. Distinguiamo inoltre forme generalizzate, che interessano tutto il corpo e forme focali, che coinvolgono solo certe parti, come gli arti o il tronco. Negli anni il soggetto distonico continuamente interessato da movimenti complessi e non finalizzati sviluppa gravi complicanze osteo-muscolari, che fissano tronco e arti in posizioni e posture viziate, che non sono più risolvibili anche riducendo i movimenti involontari.
Qual è l’origine e come si sviluppa la malattia?
Sebbene appare chiaro che la comparsa della sintomatologia distonica consegua a danni più o meno precoci nell’attività dei neuroni dei circuiti cerebrali più profondi (detti circuiti pallido-talamo-corticali), l’esatta relazione tra tali anomalie e le manifestazioni della malattia rimane non chiara. Gli studi non hanno ancora definito un meccanismo fisiopatologico univoco. Le alterazioni dei circuiti tra corteccia e nuclei profondi sono comunque molto variabili e dipendono molto dal tipo di malattia alla base della sindrome distonica. Talvolta i centri nervosi coinvolti sono iperattivi, cioè funzionano di più rispetto al soggetto normale, talvolta mostrano invece un’ipoattivazione, cioè sono meno attivi, con alcune similitudini rispetto alla malattia di Parkinson.
Quali terapie sono possibili?
L’avvento della neurochirurgia stereotassica funzionale ha segnato un momento fondamentale anche per il trattamento delle distonie. Fino al 1990 le procedure stereotassiche per trattare le forme distoniche avevano come target prevalente un centro, il “globo pallido” (Gpi), dove si praticavano lesioni. L’intervento era detto “pallidotomia” ed era irreversibile e rischioso. In seguito è stata introdotta la DBS anche per il trattamento delle sindromi distoniche. Nell’arco di pochi anni la DBS si è largamente diffusa ed il centro nervoso interessato è rimasto lo stesso. La terapia farmacologica non ha dato grossi benefici, sono state impiegate sostanza generiche, attive sul sistema nervoso centrale, già usate in molte altre malattie nervose. In altre parole, una sostanza come era stata la “L-Dopa” per la malattia di Parkinson, utile nella stessa misura per il trattamento delle distonie, non è stata mai individuata. Molte forme sono resistenti alla terapia medica e si instaurano terapie croniche soltanto sintomatiche, peraltro non scevre di effetti collaterali.
Quali risultati può ottenere la DBS? Quali sono i rischi?
Gli eccellenti risultati ottenuti nel trattamento della distonia generalizzata idiopatica resistente alla terapia medica ed anche di altre forme più focali, come quelle cervicali, ha consolidato la procedura di DBS del globo pallido come trattamento di scelta. Nelle casistiche maggiori vengono descritti miglioramenti dei punteggi nelle scale di valutazione della distonia di più dell’80 %. Tale valore, riferito a tutti i soggetti operati, si innalza al 90,3 % in certi sottogruppi di distonie, come la PGD (Primary Generalized Dystonia)-DYT1 positiva, cioè positiva per un ben preciso gene nel cromosoma 9, che è stato isolato e codificato. Ciò supporta la validità della tecnica chirurgica e del target in tutte le forme idiopatiche e soprattutto nelle forme a trasmissione ereditaria.
Nelle distonie mobili, che interessano soprattutto gli arti in movimento, il miglioramento clinico postoperatorio può essere precoce, fin dai primi giorni. Le distonie fisse invece, a carico del tronco e a riposo, presentano un miglioramento più tardivo, anche dopo mesi dall’intervento. Comunque, per valutare i risultati clinici dopo l’intervento è necessario attendere diversi mesi. Infatti anche se molti autori hanno descritto un decremento evidente dei movimenti involontari anormali subito dopo l’accensione dello stimolatore, il beneficio sulle posture e sull’aspetto funzionale globale si può valutare solo a distanza.
Dopo l’impianto è anche possibile intervenire sulle deformità secondarie dell’apparato muscolo-scheletrico, talora molto deturpanti e che, come già detto, spesso sono irreversibili anche a fronte del miglioramento neurologico ottenuto. Questo aspetto depone a favore di una necessaria precocità dell’intervento chirurgico. La procedura deve essere considerata fin dalle fasi iniziali della malattia, specie nei bambini, per i buoni risultati clinici ottenibili nella maggior parte dei pazienti, per la mancanza di un’alternativa medica appropriata, per la caratteristica di progressione nel tempo della malattia e per le gravi complicanze ortopediche che determina. Il paziente prima di essere operato viene studiato dal neurologo in modo approfondito. Esistono dei protocolli valutativi pre- e post- chirurgici molto dettagliati (valutazione clinica, neurofisiologica e molecolare), che analizzano tutti gli aspetti delle vita quotidiana del paziente. Il trattamento della distonia ad esordio precoce in età evolutiva è infatti qualcosa che coinvolge attivamente tutta la famiglia. Per quanto riguarda i rischi chirurgici specifici si rimanda alle considerazioni fatte per la chirurgia della malattia di Parkinson, inerenti le infezioni dei materiali impiantati e le emorragie cerebrali.
Qual è lo studio pre-chirurgico di selezione a cui tutti i soggetti devono sottoporsi?
1 - Esame neurologico finalizzato a valutare il controllo del capo, il controllo posturale, la persistenza di riflessi, la presenza di schemi patologici, il grado di ipertonia, l’eventuale presenza di clonie, la selettività dei movimenti, i passaggi posturali, le reazioni di equilibrio, la presa ecc.
2 - Studio neurofisiologico mediante EEG-poligrafia in corso di veglia e di sonno spontaneo con attivazioni di routine, BAEP, PESS e in casi selezionati ENG, EMG.
3 - Studio di neuroimaging mediante NMR dell’encefalo. Nella maggioranza dei casi l’esame NMR è negativo.
4 - Studio biochimico del sangue per valutare alcune sostanze che sono diagnostiche per certe malattie neurologiche. Oltre a una normale routine ematochimica vengono dosate CPK, LDH, lattato, ceruloplasmina, cupremia, aldolasi, uricemia. In casi selezionati si eseguono anche il dosaggio degli acidi organici urinari e delle acil-carnitine urinarie, acantociti, alfafetoproteina, IgA, IgE, enzimi lisosomiali (arisulfatasi A, esosaminidasi A e B, beta galattosidasi), dosaggio della neopterina in linfoblasti.
5 - Studio molecolare per l’identificazione dei geni e delle mutazioni. La più nota è la delezione GAG nel gene DYT-1 responsabile della distonia idiopatica generalizzata di torsione (Oppenheim dystonia), già precedentemente menzionata. Inoltre, in casi selezionati (pazienti per i quali, sulla base dei dati clinici, strumentali e bioumorali, venga sospettata una possibile patologia mitocondriale) vengono ricercate le mutazioni del mtDNA più frequentemente coinvolte nei quadri encefalopatici
6 - Scala per la valutazione della distonia (Burke-Fahn-Mardsen, BFM). La scala serve a valutare i movimenti distonici e le posture distoniche. Comprende l’osservazione diretta del soggetto e appositi video (osservazione a riposo, movimenti involontari, lettura e fonazione, prove di scrittura, prove di abilità manuale, ecc). La scala conferisce un punteggio massimo di 120. Inoltre si eseguono scale di disabilità per linguaggio, scrittura, alimentazione, nutrizione, igiene, deambulazione, vestirsi.
7 - Intervista sulla disabilità riguardante l’autonomia nella cura di sé, somministrata ai genitori del paziente.
8 - Profilo neuropsicologico con valutazione delle funzioni cognitive generali, delle funzioni visuospaziali, esecutive, mnesiche e percettive.
9 - Valutazione logopedica adeguata e bilancio della capacità di esprimersi verbalmente.
Tutti i pazienti vengono sottoposti ad un tentativo farmacologico con L-Dopa, per poter identificare eventuali forme di distonia responsiva a questa sostanza, che sono comunque relativamente rare. Viene inoltre intrapreso un trattamento farmacologico mediante l’uso isolato oppure l’associazione di L-Dopa, Anticolinergici, Tetrabenazina o Baclofen.
Esistono criteri di inclusione per la DBS?
In base al protocollo di studio preoperatorio descritto, possiamo schematizzarli come segue:
1 - età superiore a 7 anni
2 - NMR cerebrale negativa
3 - profilo cognitivo-psichiatrico normale e adeguato screening preoperatorio
4 - assenza di risposta clinicamente rilevante al trattamento farmacologico
5 - disabilità severa, fortemente condizionata dalla componente distonica (BFM > 50-60)
Come si svolge l’intervento neurochirurgico?
La procedura di DBS, quando viene eseguita in paziente adulti, prevede il paziente sveglio e la microregistrazione intraoperatoria. La metodica è analoga al trattamento della malattia di Parkinson. Nei bambini invece si impiega un’anestesia generale con o senza microregistrazione (
In età pediatrica si impianta tutto lo stesso giorno, sia gli elettrocateteri cerebrali, che lo stimolatore definitivo, mentre negli adulti si praticano due interventi distinti, come nel Parkinson. Dopo una settimana dall’impianto si procede all’accensione dello stimolatore ad alta frequenza, con intensità maggiori rispetto al Parkinson, che vegono progressivamente incrementate a seconda dei risultati clinici.
Come viene seguito il paziente operato? In questa indicazione chirurgica relativamente nuova, alla luce dell’attuale esperienza quali considerazioni si possono fare?
Dopo la dimissione dalla Neurochirurgia i pazienti vengono seguiti clinicamente dal neurologo con scadenze settimanali per il primo mese ed, in seguito con controlli sequenziali a 1, 3, 6, 12 mesi dall’intervento. Vengono applicati gli stessi test valutativi somministrati in fase pre intervento. Contemporaneamente all’accensione dello stimolatore si procede al trattamento riabilitativo (logopedia, terapia occupazionale, fisiochinesiterapia) volto a modellare i nuovi schemi motori esistenti, sulla base di maggiori capacità di controllo del movimento e della postura.
La selezione dei pazienti distonici è fondamentale per ottenere il maggior beneficio clinico e soggettivo da un intervento di DBS, minimizzando gli effetti avversi, sia nelle forme primarie che secondarie. Vi è un crescente consenso nel ritenere che l’intervento dovrebbe pertanto essere proposto come trattamento di prima linea nei casi di distonia generalizzata primaria e secondaria non responsiva alla terapia farmacologica.